Migration of GnRH neurones
(The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Suzy D. C. Bianco and Ursula B. Kaiser. Aug 25, 2009. Nature.com With permission)
This diagram shows the site of activity of some of the more common genes associated with Kallmann syndrome and congenital hypogonadism.
They can be separated into two broad categories:
Movement of the GnRH releasing neurones from the nasal placode into the hypothalamus during the first 10 to 14 weeks of development.
or
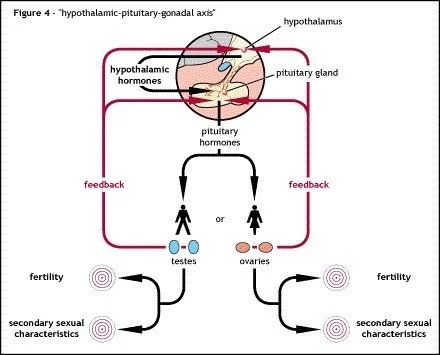
The ability of the hypothalamus to release GnRH in a pulsatile manner so that the pituitary gland is able to release LH in the correct manner to have an influenced on the gonads (ovaries or testes) and induce steroid hormone production (oestrogen / progesterone or testosterone).
The migration of the GnRH releasing neurones is normally along the same pathway as the olfactory (sense of smell) neurones. If the GnRH releasing neurones are blocked, so are the olfactory neurones which can result in the anosmia seen in Kallmann syndrome but not CHH.
The release of GnRH in a pulsatile manner is tightly controlled by external factors, including a protein called Kisspeptin. There are a number of genes that are involved in these control mechanisms or the ability of the pituitary gland to recognise GnRH via receptors. Any defect in these genes can lead to a problem in the release of GnRH which can cause CHH.
It should be noted that more than one gene or gene(s) can be affected. This makes inheritance prediction very difficult, it can also produce a range of severity of symptoms within families, even siblings, depending on which genes have been affected and at what stage in the development process the genes are supposed to operate.